ChimeraX Matchmaker
Dive into the world of “Matchmaker”, an essential ChimeraX extension for protein structure-function research. This blog post unveils its intricacies, guiding you through both its fundamentals and advanced features. Learn how Matchmaker seamlessly superimposes protein and nucleic acid structures, offering insights into the complex realm of molecular biology.
Learning Objectives:
At the end of this lesson, you should be able to:
- Understand Matchmaker’s role in protein structure research
- Learn the key steps of structure superimposition
- Explore chain pairing options in Matchmaker
- Understand sequence alignment and scoring settings
- Gain insights into the fitting process
1. What is Matchmaker?
In the field of protein structure-function research, a common task is to analyze different protein structures and understand how their shapes relate to their functions. To do this effectively, we need to compare these structures and see how they fit together. This comparison is made possible by aligning the structures in a way that makes sense, ensuring that we can draw meaningful insights from the process.
Matchmaker is the ChimeraX extension that enables integrated sequence-structure analysis. The functionality of this extension can be accessed either through the graphical user interface (UI) or the command line interface (using the matchmaker command). To access the UI, users can click on the menu option Tools -> Structure Analysis -> Matchmaker.
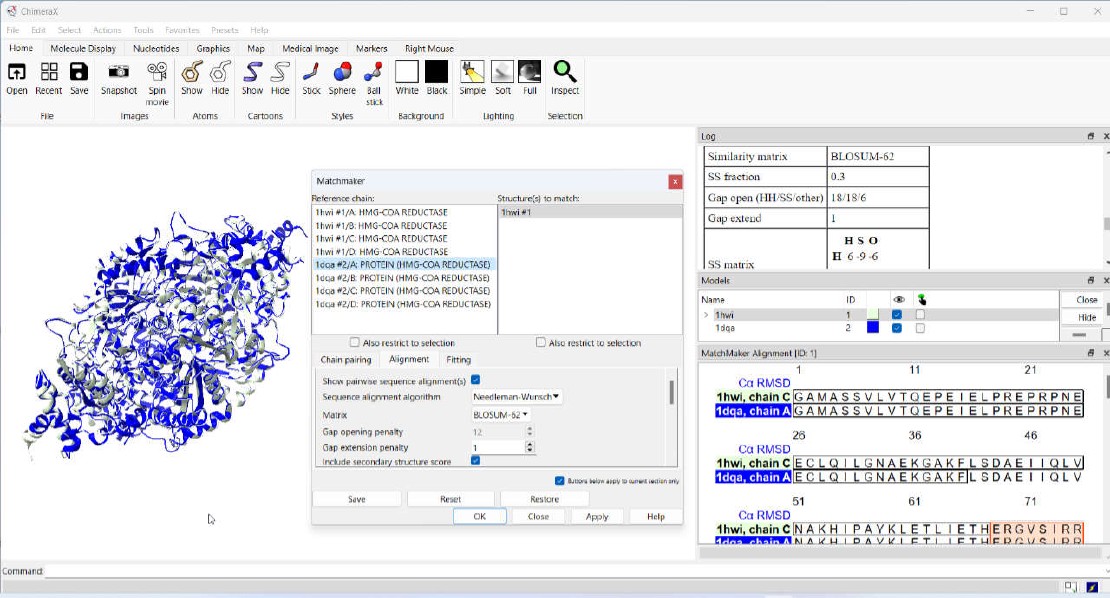
The top portion of the Matchmaker UI is divided into two halves; the left half shows the reference structure and the right half shows one or more structures to match. The bottom part of the UI is divided into 3 tabs; Chain pairing, Alignment, and Fitting. The content displayed in the upper two panels is dictated by the configurations within the Chain pairing tab.
2. How Does Matchmaker Superimpose Structures?
The matchmaker tool is capable of superimposing protein or nucleic acid structures. The process involves 3 basic steps.
- Creating a pairwise sequence alignment.
- Aligning the residues based on sequence, residue type, and secondary structure information.
- Performing a least squares fit by calculating the distances between equivalent atomic positions in the two structures.
3. Matchmaker Chain Pairing
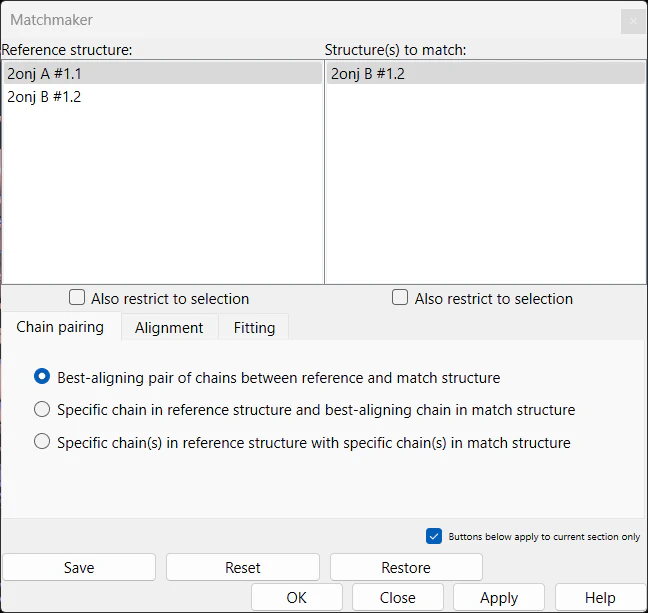
The Chain pairing tab dictates the choices of chains available for matching. In this article, when we mention structures," we are referring to three-dimensional models that consist of multiple chains. The first option (initial default option), Best-aligning pair of chains between reference and match structures, allows you to choose one reference structure and one or more match structures. The algorithm will select the pairs of chains (between reference and match structures) with the highest sequence alignment scores. Specific chain in reference structure and best-aligning chain in match structure lets you select a chain from the reference structure and one or more match structures. The algorithm will pick the best matching chains from the match structures.

The third option, Specific chain(s) in reference structure with specific chain(s) in match structure allows you to pick chains from both reference and match structures.
Matchmaker also provides a way to restrict the superposition to a selected set of residues from reference and match structures. This can be achieved by checking the checkboxes,Also restrict to selection under reference and match structures.
However, such restrictions are generally not recommended as full-length alignments tend to be of higher quality. One example where restrictions might be useful is when you want to force the N-terminal domain-aligned chains to align on the C-terminal domain. In any case, all residues (both selected and unselected) of the alignment will be shown on the sequence viewer. The unselected residues are added back in as filler residues without any regard to alignment.
4. Matchmaker Alignment
ChimeraX Matchmaker’s alignment score may incorporate contributions from the secondary structure. The setting Include secondary structure score is enabled by default. The default alignment score includes a 30% contribution from the secondary structure score.
When this setting is disabled, the section titled Secondary structure scoring disappears from the alignment tab. This change in settings also activates the initially grayed-out Gap opening penalty parameter. This particular parameter is ignored in the presence of secondary structure scoring. A secondary structure-specific gap opening penalty is used instead.
The settings in the alignment panel can be divided into two groups: those related to sequence alignment and those associated with secondary structure scoring.
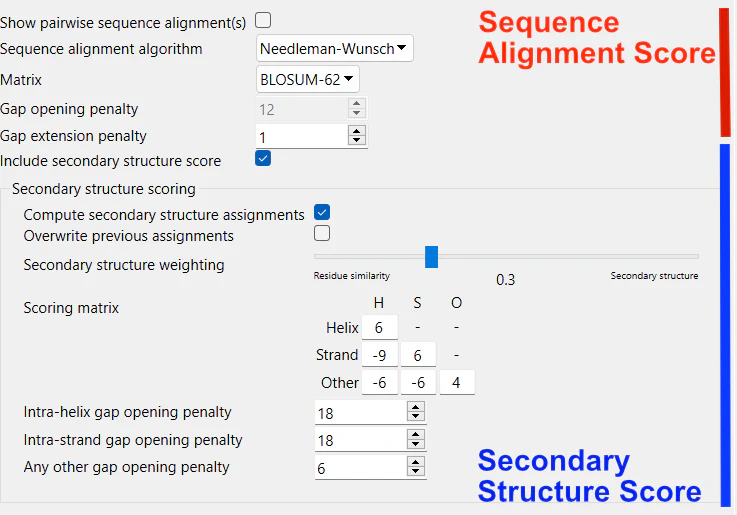
4.1. Sequence Alignment Settings
-
Show pairwise sequence alignment(s): By default, this is disabled. When enabled, each pair-wise alignment (between reference-match pairs)will be shown in a separate window.
-
Sequence alignment algorithm: What sequence alignment algorithm is to be used? The initial default is the global sequence alignment algorithm, Needleman-Wunsch. Smith-Waterman algorithm is also available for local alignments.
-
Matrix: Which substitution matrix to use for calculating the residue similarity? The initial default is BLOSUM-62.
-
Gap Opening Penalty: The penalty for opening a gap in the alignment. Increasing this value makes the gaps less frequent. Initial defalut is 12.
-
Gap Extension Penalty: The penalty for extending a gap by one residue. Increasing this value will make the gaps shorter. The initial default is 1.
4.2. Secondary Structure Scoring Settings
-
Compute Secondary Structure Assignments: Decide whether to employ the DSSP algorithm for helix and strand identification. DSSP is the industry-standard technique for assigning secondary structure to a protein’s amino acids based on atomic-resolution coordinates. This setting is activated by default.
-
Overwrite Previous Assignments: Choose whether to replace existing secondary structure assignments with newly calculated ones. Otherwise, the new assignments serve a temporary purpose, mainly for superposition.
-
Secondary Structure Weighting (Initial Default: 0.30): Adjust the fractional weight ‘f’ governing the contribution of secondary structure to the overall score. The residue similarity contribution is weighted as (1 - f). For instance, a value of 0.3 yields: Total score = 0.30(secondary structure score) + 0.70(residue similarity score) – gap penalties. Please note that setting the slider to 0.0 does not equate to turning off secondary structure scoring. When this option is active, secondary structure-specific gap opening penalties are applied, regardless of the slider’s position.
-
Customize Secondary Structure Scoring: Tailor the values within the secondary structure scoring matrix for all possible combinations of H (helix), S (strand), and O (other), as well as the specific gap opening penalties for secondary structure elements (intra-helix, intra-strand, and any other).
5. Matchmaker Fitting
Fitting of residues involves a least squares fit. The distance measurements used in the least squares fit are calculated with one point (atomic position of one atom) per residue. When fitting proteins, alpha carbons (Ca) are used for the distance calculations.
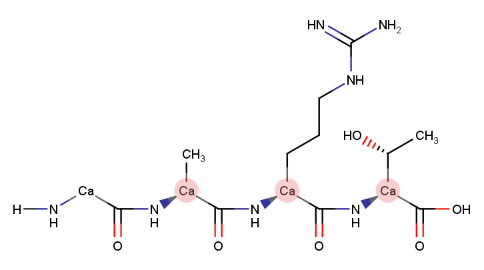
For nucleic acids, C4’ atoms are utilized. When the structures lack C4’ atoms (low-resolution structures with P traces) P atoms are employed.
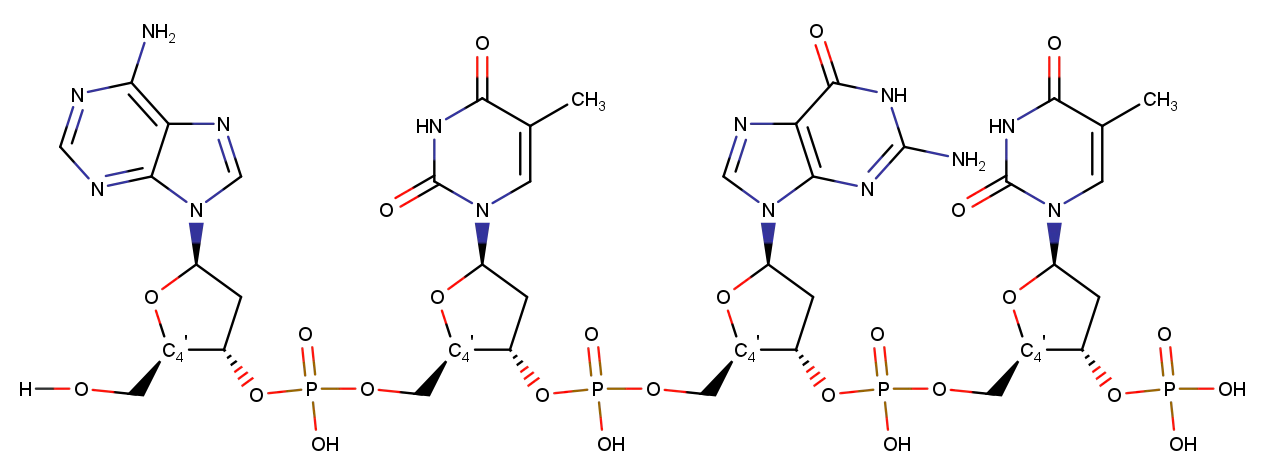
During the process of fitting protein and nucleic acid structures, it is also possible to employ custom atom types in conjunction with the align command.
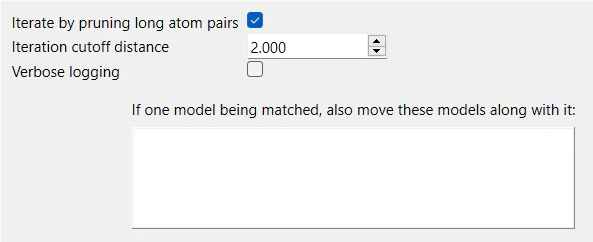
The Fitting tab includes several settings that deal with:
- Pruning
- Logging
- Moving models
5.1. Pruning
Pruning is the process of removing far-apart residue pairs from the superimposition. It involves gradually removing distant atom pairs from a list used to align two structures. This process doesn’t change the initial alignment of the sequences but limits which parts of the alignment are considered for the final alignment.
Normally, all columns with both sequences (no gaps) are used. However, in each iteration, atom pairs are removed until no pairs are farther apart than a specified distance.
The removed pairs are either the 10% farthest apart or the 50% farthest apart beyond a certain cutoff, whichever is fewer. This iterative approach helps exclude regions with different shapes, like flexible loops, and results in a better fit for the most similar core regions. The initial default distance for this cutoff is 2.0 Å.
Pruning is turned on by default. The two settings, Iterate by pruning long atom pairs and Iteration cutoff distance that go hand-in-hand determines what residues are removed from the superimposition. When the check box for the first setting is checked the user can change the cutoff distance. Otherwise, the second setting is grayed off.
5.2. Logging
The setting Verbose logging controls the amount of information displayed on the log panel. By default, this checkbox is turned off. When it is turned off, you get a summary of the superimposition on the log panel. The entire list of residues used in the superimposition, their residue names, positions, and what residues were excluded can be viewed with verbose logging.
5.3. Moving models
This setting controls how other models on the workspace should be moved with the superimposed models. It is particularly valuable when dealing with structures that consist of multiple models, such as those found in NMR studies.
6. Final Thoughts
Matchmaker, a crucial part of ChimeraX, makes protein research easier. It helps align protein and nucleic acid structures, simplifying molecular biology exploration. From basics to advanced features, Matchmaker guides users through structure alignment, chain pairing, and alignment settings.
Whether you use the UI or command line, Matchmaker streamlines sequence-structure analysis, making it easier to understand relationships between different protein structures. Dive in to understand molecular structures and their functions more deeply with Matchmaker.
Related Posts

ChimeraX Command Line Interface (CLI)
Discover the capabilities of ChimeraX’s Command Line Interface (CLI) for seamless control of its numerous functions. Gain proficiency in accessing the CLI, browsing through command history, and harnessing the flexible “open” command for handling both local and online data.
Read more
An Introduction to ChimeraX User Interface
Welcome to our blog post on ChimeraX User Interface. ChimeraX is a powerful molecular visualization and analysis software widely used by researchers in the field of structural biology.
Read more
Navigating PDB: Legacy Format to PDBx/mmCIF
Discover the fascinating journey of protein data storage since 1971. From the legacy PDB format, designed for punched cards, to the modern PDBx/mmCIF format, built to accommodate complex macromolecular data.
Read more